С учетом продолжающегося развития высокопроизводительных секвенирующих машин и беспрецедентного развития современных биоинформационных инструментов целевая цель секвенирования отдельных геномов живого организма стоимостью 1000 долларов КАЖДЫЙ представляется реально осуществимой в ближайшем будущем. В относительно короткие сроки, начиная с 2005 года, технологии HT-NGS революционизируют исследования генома человека и животных путем анализа иммунопреципитации хроматина в сочетании с микрочипом ДНК (ChIP-chip) или секвенированием (ChIP-seq), секвенированием РНК (RNA-seq), генотипированием всего генома, широкой структурной вариацией генома, сборкой de novo и повторной сборкой генома, обнаружением мутаций и скринингом носителей, обнаружением наследственных нарушений и сложных заболеваний человека, подготовкой библиотеки ДНК, парными концами и геномными захватами, секвенированием генома.митохондриальный геном и личная геномика. В этом обзоре мы рассмотрели важные особенности HT-NGS, такие как секвенсоры ДНК первого поколения, рождение HT-NGS, платформы HT-NGS второго поколения, платформы HT-NGS третьего поколения: включая одномолекулярные Heliscope ™ , SMRT ™ и RNAP секвенсоры, Nanopore, Archon Genomics X PRIZE foundation, сравнение второй и третьей платформ HT-NGS, приложения, достижения и будущие перспективы технологий секвенирования в исследованиях генома человека и животных.
Завершение первых проектов генома человека (Yamey 2000) было лишь началом современной эры секвенирования ДНК, которая привела к дальнейшим изобретениям, усовершенствованным разработкам в направлении новых передовых стратегий высокопроизводительного секвенирования ДНК, так называемого “высокопроизводительного секвенирования следующего поколения” (HT-NGS). Эти разработанные стратегии HT-NGS были направлены на удовлетворение наших ожидаемых будущих потребностей в секвенировании пропускной способности и стоимости таким образом, чтобы обеспечить его потенциальное множество текущих и будущих применений в геномных исследованиях млекопитающих. Кроме того, в этих передовых лабораторных методологиях сфера применения инструментов биоинформатики нового поколения стала еще более важной предпосылкой для обеспечения дальнейшего стратегического развития и улучшения выходных результатов. HT-NGS-одна из главных проблем современных геномных исследований. Для будущего направления нам нужна глубокая информация о последовательности генома и анализ для большинства млекопитающих, включая человека, чтобы полностью понять геномные вариации экономических признаков, генетическую восприимчивость к болезням и фармакогеномику лекарственного ответа.
Ведущие исследовательские центры генома и ученые публично признали, что это основные цели, способствующие проведению исследований в области геномики в следующем десятилетии. Национальный исследовательский институт генома человека (NHGRI) повторил эту потребность в своем видении геномных исследований (Collins et al., 2003). NHGRI классифицировала новые подходы к секвенированию на те, которые предлагают краткосрочные и революционные преимущества с 100-кратным снижением затрат на одну базовую пару (bp) в течение следующих пяти лет. Чтобы продлить ближайшую перспективу, то есть в течение следующих 5-10 лет, революционные преимущества должны продвинуть область с 10 000-кратным снижением затрат на одну базовую пару, что, в свою очередь, позволит достичь “генома в 1000 долларов США”.
2011 год отмечается как 10-летие со дня первого секвенирования генома человека (www.nature.com/natureconferences/hg10years/index.html). За этот период были достигнуты огромные успехи в области расшифровки генома человека, технического прогресса новой эры применения генома человека, в направлении персонализированных геномов и открытия редких вариантов, использования секвенирования генома для воздействия на исследования рака и эволюцию млекопитающих и структуру популяции. В последнее десятилетие произошла революция в области исследований геномики человека. Сегодня принимается более глобальный подход, который не только дал начало области системной биологии, но и затронул все области биологических и медицинских исследований, а также сблизил их и размыл границы, которые ранее определяли их как отдельные дисциплины исследований. Горизонты и ожидания расширились благодаря технологическим достижениям в области геномики, особенно HT-NGS и его широкому спектру применений, таких как: иммунопреципитация хроматина в сочетании с микрочипом ДНК (ChIP-chip) или секвенирование (ChIP-seq), секвенирование РНК (RNA-seq), генотипирование всего генома, сборка de novo и повторная сборка генома, широкая структурная вариация генома, обнаружение мутаций и скрининг носителей, обнаружение наследственных нарушений и сложных заболеваний человека, подготовка библиотеки ДНК, парные концы и геномные захваты., секвенирование митохондриального генома и персональная геномика (подробное описание см. в таблице 2).
Помимо развития методов секвенирования, прошедшее десятилетие запомнится как десятилетие исследований генома. Со времени публикации первых составных геномов человека (Lander et al. 2001; Venter et al. 2001) было опубликовано много черновых геномов других организмов (www.ensembl.org/info/about/species.html). Скорость, с которой теперь можно секвенировать новые геномы, была облегчена развитием потенциальных технологий HT-NGS и методов сборки. Теперь можно собрать de novo большой геном, хороший пример может быть получен из недавней сборки генома гигантской панды (Li et al. 2010b), которая использовала только короткие считывания, обеспеченные секвенированием ДНК следующего поколения.
Секвенсоры ДНК первого поколения
Исторически сложилось так, что в 1975 году Сэнгер ввел концепцию метода секвенирования ДНК в своей пионерской Кроновской лекции (Sanger 1975), а позже опубликовал быстрый метод определения последовательностей в ДНК путем праймерного синтеза с ДНК-полимеразой (Sanger and Coulson 1975). В 1977 году были опубликованы две знаковые статьи по секвенированию ДНК, а именно метод ферментативного секвенирования дидеокси-ДНК Фредерика Сэнгера, основанный на цепочечных аналогах дидеоксинуклеотидов (Sanger et al. 1977) и метод химического секвенирования ДНК Аллана Максама и Уолтера Гилберта, в котором терминально меченые фрагменты ДНК химически расщеплялись на определенных основаниях и отделялись гель-электрофорезом (Maxam and Gilbert 1977). Эти две выдающиеся элитные лаборатории были ответственны за внедрение первых автоматизированных секвенсоров ДНК во главе с Caltech (Smith et al. 1986), которые впоследствии были коммерциализированы Applied Biosystems (ABI), Европейской лабораторией молекулярной биологии (EMBL) (Ansorge et al. 1986, 1987) и Pharmacia-Amersham, позже General Electric (GE) healthcare. Это усовершенствование и коммерциализация метода секвенирования привели к его широкому распространению в мировом исследовательском сообществе.
В первом автоматизированном оборудовании для флуоресцентного секвенирования ДНК был секвенирован полный локус гена гипоксантин-гуанинфосфорибозилтрансферазы (HPRT), впервые используя метод парного секвенирования (Edwards et al., 1990). В 1996 году компания ABI представила первый коммерческий секвенсор ДНК, в котором использовался слябовый гель-электрофорез с помощью призмы ABI Prism 310. Через два года значительная трудоемкость заливки слябовых гелей была заменена автоматизированной перегрузкой капилляров полимерной матрицей ABI Prism 3700 с 96 капиллярами. Этот автоматизированный секвенсор ДНК был успешно использован при секвенировании первого генома человека в 2003 году с учетом 13-летних усилий консорциума проекта "геном человека" и предполагаемой стоимостью 2,7 миллиарда долларов. В последующие годы была достигнута еще одна веха-секвенирование ДНК первого небольшого генома фага (5386 оснований в длину) и секвенирование генома человека длиной до 3 миллиардов оснований (Lander et al., 2001; Venter et al., 2001). Примечательно, что такой прогресс был достигнут с использованием методов, являющихся усовершенствованиями основного метода "дидеоксии", введенного Сэнгером в 1977 году.
Рождение HT-NGS
В 2000 году Джонатан Ротберг основал компанию 454 Life Sciences, которая в дальнейшем разработала первую коммерчески доступную платформу NGS-GS 20. Инструмент GS был представлен в 2005 году и разработан компанией 454 Life Sciences (www.454.com), как первая система NGS на рынке. Разработанная методика была успешно валидирована путем комбинирования одномолекулярной эмульсионной ПЦР с пиросеквенированием (процедура дробового секвенирования) всего 580 069 бп генома Mycoplasma genitalia с охватом 96% и точностью 99,96% в одном прогоне GS 20 (Margulies et al. 2005).
В последующие годы Roche applied science приобрела 454 Life sciences и расширила новую версию прибора 454, то есть GS FLX titanium. Разделяя один и тот же технологический принцип как в GS 20, так и в GS FLX titanium, проточная ячейка называется пластиной "пикотитровой скважины", которая изготавливается из плавленого волоконно-оптического пучка. На отдельном фронте одномолекулярная ПЦР в микрокомпонентах, состоящих из эмульсий вода-в-масле, была также разработана платформой Roche HT-NGS (Tawfik and Griffiths 1998). В общем, принцип техники пиросеквенирования основан на “секвенировании синтезом”. Он отличается от секвенирования Сэнгера тем, что зависит от обнаружения высвобождения пирофосфата при инкорпорации нуклеотидов, а не от прекращения цепи дидеоксинуклеотидами. Методика была разработана совместными усилиями шведской группы (команды M. Ronaghi, M. Uhlen и P. Nyren) в Стокгольме (Ronaghi et al., 1996). Они впервые описали метод секвенирования, основанный на хемилюминесцентном обнаружении пирофосфата, высвобождаемого при инкорпорации полимеразно-опосредованного дезоксинуклеозидтрифосфата (dNTP) (Nyren et al. 1993, Nyren 2007), и секвенировании ДНК в реальном времени с использованием этого высвобождения пирофосфата (Ronaghi et al. 1998). При пиросеквенировании синтез ДНК осуществляется в рамках сложной реакции, включающей ферменты АТФ-сульфурилазы и люциферазы, а также субстраты аденозин-5' - фосфосульфата и люциферина таким образом, что пирофосфатная группа высвобождается при добавлении нуклеотида, что приводит к образованию детектируемого света.
Методы HT-NGS, которые являются новыми возможностями и оказывают большое влияние на исследования геномики млекопитающих, были выбраны в качестве методов года в 2007 году (Schuster et al. 2008). Однако путь к признанию этих новых технологий был нелегким. Первый шаг метода HT-NGS состоял в обнаружении следующего добавленного флуоресцентно меченого основания (обратимого терминатора) в растущей цепи ДНК с помощью чувствительной ПЗС-камеры. Это было выполнено на большом количестве образцов ДНК параллельно, прикрепленных либо к плоской опоре, либо к шарикам, на ДНК-чипах, минимизируя объемы реакции в миниатюрной микросистеме. На следующем этапе терминатор превращался в стандартный нуклеотид, а краситель удалялся. Этот цикл и процесс повторялись, чтобы определить следующую базу в последовательности. Принцип, описанный в этом приложении, отчасти очень похож на тот, который используется сегодня в так называемых устройствах следующего поколения, коммерциализируемых Roche, Illumina-Solexa, ABI, Helicos и другими компаниями.
Принцип HT-NGS включает в себя молекулы ДНК, которые секвенируются массивно-параллельным образом в проточной ячейке (Mardis 2008a, b; Metzker 2010). Секвенирование проводится либо в пошаговом итерационном процессе, либо в непрерывном режиме реального времени. Благодаря этому очень параллельному процессу каждая клональная матрица или отдельная молекула “индивидуально” секвенируется и может быть подсчитана среди всех генерируемых последовательностей. Высокая пропускная способность комбинации качественной и количественной информации о последовательностях позволила провести продвинутый анализ генома, который ранее был либо технически невозможен, либо стоил непомерно дорого.
Платформы второго поколения HT-NGS
Платформы второго поколения HT-NGS могут генерировать около пятисот миллионов баз необработанной последовательности (Roche) до миллиардов баз за один запуск (Illumina, SOLiD). Эти новые методы основаны на параллельном циклическом опросе последовательностей из пространственно разделенных клональных ампликонов (26 мкм масляно-водной эмульсионной бусины [Roche: pyrosequencing chemistry], 1 мкм клональной бусины [SOLiD: sequencing by sequential ligation of oligonucleotide probes], клонального моста [Illumina: sequencing by reversible dye terminators]). В настоящее время эти (упомянутые выше) три ведущие платформы второго поколения HT-NGS являются коммерчески доступными, и гонка за более дополнительными платформами постоянно находится на горизонте (для всесторонних обзоров полных лабораторных методов, технических аспектов подготовки образцов и последующего анализа данных секвенирования Roche, Illumina, SOLiD platforms см.: Mardis 2008a, b, 2009, 2010; Metzker 2010).
В 2008 году Национальный исследовательский институт генома человека США (NHGRI) инициировал финансирование ряда проектов в рамках своей революционной программы технологий секвенирования генома и нацелился на достижение своей целевой цели секвенирования генома человека за 1000 долларов или меньше (http://www.genome.gov/27527585). Недавно, в декабре 2010 года, консорциум NHGRI опубликовал наиболее полную карту генетической изменчивости человека с использованием технологий секвенирования ДНК следующего поколения для систематической характеристики генетических различий между 179 особями из четырех популяций и 697 особями из семи популяций в трех пилотных исследованиях (Durbin et al. 2010). Эти пилотные исследования “проекта 1000 геномов” заложили критическую основу для изучения генетической изменчивости человека и были направлены на создание всеобъемлющей, общедоступной карты генетической изменчивости, которая в конечном итоге соберет последовательность от 2500 человек из различных популяций по всему миру и послужит основой для будущих генетических исследований (http://www.genome.gov/27541917).
Платформы HT-NGS третьего поколения
В ранее обсуждавшихся платформах второго поколения HT-NGS принцип был основан на эмульсионной ПЦР-амплификации фрагментов ДНК, чтобы сделать световой сигнал достаточно сильным для надежного обнаружения базы ПЗС-камерами. Хотя ПЦР-амплификация произвела революцию в анализе ДНК, но в некоторых случаях она может вводить ошибки базовой последовательности или отдавать предпочтение определенным последовательностям перед другими, тем самым изменяя относительную частоту и обилие различных фрагментов ДНК, существовавших до амплификации. Чтобы преодолеть это, конечная миниатюризация в наноразмерном масштабе и минимальное использование биохимических веществ были бы достижимы, если бы последовательность могла быть определена непосредственно из одной молекулы ДНК, без необходимости ПЦР-амплификации и ее потенциала для искажения уровней изобилия. Это секвенирование из одной молекулы ДНК теперь называется “третье поколение технологии HT-NGS” (Schadt et al., 2010). Концепция секвенирования путем синтеза без предварительного этапа амплификации, то есть секвенирования одной молекулы, в настоящее время используется рядом компаний и описана ниже в Разделах. 5.1 - 5.7.
Одномолекулярный секвенсор Heliscope ™
Один из первых методов секвенирования из одной молекулы ДНК был представлен Braslavsky et al. в 2003 году и лицензирован Helicos biosciences в качестве первой коммерческой системы секвенирования одномолекулярной ДНК в 2007 году. Принцип работы секвенсора Heliscope основан на технологии “истинного одномолекулярного секвенирования” (TSMS). Технология tSMS начинается с подготовки библиотеки ДНК путем сдвига ДНК и добавления поли-(А) хвоста к генерируемым фрагментам ДНК (Ozsolak et al. 2010), с последующей гибридизацией фрагментов ДНК с поли-(Т) олигонуклеотидами, которые присоединяются к проточной клетке и одновременно секвенируются в параллельных реакциях. Цикл секвенирования состоит из расширения ДНК одним из четырех флуоресцентно меченых нуклеотидов с последующим обнаружением нуклеотидов с помощью секвенсора Heliscope. Последующее химическое расщепление флуорофоров позволяет начать следующий цикл удлинения ДНК с другого флуоресцентно меченого нуклеотида, что позволяет определить последовательность ДНК (Harris et al., 2008). Секвенсор Heliscope способен секвенировать до 28 Гб за один запуск секвенирования и занимает около 8 дней. Он может генерировать короткие считывания с максимальной длиной 55 баз. В недавней разработке Helicos объявила, что она разработала новое поколение нуклеотидов “по одному основанию за раз”, которые позволяют более точно гомополимерировать и прямое секвенирование РНК (Ozsolak and Milos 2011a, b).
Одномолекулярный секвенсор реального времени (SMRT ™ )
Принцип секвенсора SMRT основан на секвенировании одной молекулы в реальном времени методом синтеза, предусмотренным на микросхеме секвенирования, содержащей тысячи волноводов нулевой моды (ZMW). Реакция секвенирования фрагмента ДНК выполняется одной молекулой ДНК-полимеразы, которая прикреплена к нижней части каждого ZMW так, что каждая ДНК-полимераза находится в зоне обнаружения ZMW.
Во время реакции секвенирования фрагмент ДНК удлиняется ДНК-полимеразой с дНТП, которые флуоресцентно мечены (каждый нуклеотид мечен флуорофором разного цвета) на концевом фосфатном фрагменте. Последовательность ДНК определяется с помощью ПЗС - матрицы на основе флуоресцентной детекции нуклеотидов, которая выполняется до включения нуклеотидов, в то время как меченый дНТП образует родственную ассоциацию с матрицей ДНК. Импульс флуоресценции прекращается после образования фосфодиэфирной связи, что вызывает высвобождение флуорофора, который диффундирует из ЗМВ. Впоследствии инкорпорация и детекция меченых нуклеотидов позволяют определить последовательность ДНК (Levene et al. 2003; Eid et al. 2009). Секвенсор SMRT был разработан и до сих пор разрабатывается Pacific Biosciences (www.pacificbiosciences.com). Хотя инструмент SMRT недавно появился на рынке, компания утверждает, что анализатор SMRT может быть способен получать 100 Гб в час при чтении более 1000 за один запуск.
Одномолекулярный секвенсор реального времени (RNAP)
Другой подход к секвенированию одномолекулярной ДНК, то есть РНК-полимераза (RNAP), был предложен (Greenleaf and Block 2006), в котором RNAP прикрепляется к одному полистирольному шарику, в то время как дистальный конец фрагмента ДНК прикрепляется к другому шарику. Каждая бусина помещается в оптическую ловушку, и пара оптических ловушек левитирует бусины. RNAP взаимодействует с фрагментом ДНК, и транскрипционное движение RNAP вдоль шаблона изменяет длину ДНК между двумя шариками. Это приводит к смещению двух шариков, которые могут быть зарегистрированы с точностью в диапазоне Ангстрема, что приводит к одноосновному разрешению на одной молекуле ДНК. Выравнивая четыре записи смещения, каждая с более низкой концентрацией одного из четырех нуклеотидов, в роли, аналогичной праймерам, используемым в секвенировании Сэнгера и для калибровки с использованием известных последовательностей, фланкирующих неизвестный секвенированный фрагмент, можно вывести информацию о последовательности. Методика демонстрирует движение фермента нуклеиновой кислоты и очень чувствительный метод оптической ловушки, который может позволить извлекать информацию о последовательности непосредственно из одной молекулы ДНК.
Нанопористый секвенсор ДНК
В отличие от всех упомянутых выше секвенсоров ДНК, секвенирование молекулы ДНК с помощью нанопористого секвенсора ДНК не требует маркировки и обнаружения нуклеотидов. Эта методика была разработана на основе исследований транслокации ДНК через различные искусственные нанопоры. Секвенирование ДНК с помощью нанопористого прибора основано на преобразовании электрического сигнала нуклеотидов путем прохождения через нанопору, представляющую собой пору α-гемолизина, ковалентно связанную с молекулой циклодекстрина – сайтом связывания нуклеотидов. Принцип этой методики основан на модуляции ионного тока через пору по мере прохождения через нее молекулы ДНК, выявляющей характеристики и параметры (диаметр, длину и конформацию) молекулы (рис. 2). В процессе секвенирования ионный ток, проходящий через нанопору, блокируется нуклеотидом, то есть ранее отщепленным экзонуклеазой от нити ДНК, взаимодействующей с циклодекстрином. Период времени текущего блока характерен для каждого основания и позволяет определить последовательность ДНК (Astier et al. 2006; Rusk 2009). Однако дальнейшие усовершенствования и модификации в этой методике, например увеличение числа параметров, измеряемых во время транслокации ДНК, позволяющей получить разрешение с одним основанием, могут привести к быстрому секвенированию ДНК на основе нанопор.
Платформы секвенсора одномолекулярной ДНК в реальном времени разработанные компанией VisiGen Biotechnologies
Вестигенские биотехнологии (www.visigenbio.com) ввели специально сконструированную ДНК-полимеразу, которая действовала как "датчик реального времени" для модифицированных нуклеотидов с донорным флуоресцентным красителем и инкорпорировалась вблизи активного сайта, участвующего в отборе нуклеотидов в процессе синтеза (рис. 2). Все четыре нуклеотида, которые должны были быть интегрированы, были модифицированы, каждый с различным акцепторным красителем. Во время синтеза, когда правильный нуклеотид найден, он выбран и введен в активный сайт фермента, а метка донорного красителя в полимеразе оказалась в непосредственной близости с акцепторным красителем на нуклеотидах, и энергия была передана от донора к акцепторному красителю, что привело к флуоресцентному резонансному переносу энергии (FRET) светового сигнала (Selvin 2000). Частота этого сигнала менялась в зависимости от метки, заложенной в нуклеотидах, так что путем регистрации частот испускаемых сигналов лада можно было определить базовые последовательности, со скоростью, с которой полимераза может интегрировать нуклеотиды в процессе синтеза (обычно несколько сотен в секунду). Акцепторный флуорофор удаляется во время инкорпорации нуклеотидов, что гарантирует отсутствие модификаций ДНК, которые могли бы замедлить полимеразу во время синтеза. В настоящее время компания работает над своей первой версией инструмента, который может генерировать около 4 Гб данных в день. Одномолекулярный подход не требует клонирования и амплификации, что исключает значительную часть затрат по сравнению с современными технологиями. Кроме того, ожидается, что длина считывания для прибора составит около 1 кб, что больше, чем у любой текущей платформы.
Мультиплексная технология polony
Руководит частным проектом personal genome project (PGP) и возглавляет его проф. Исследовательская группа Г. Черча (www.personalgenomes.org), разработал и внедрил мультиплексную технологию полония (Mitra et al. 2003; Shendure et al. 2005). В этом методе несколько сотен шаблонов секвенирования наносятся на тонкие слои агарозы и последовательности определяются параллельно. Этот метод представляет собой увеличение на несколько порядков количества образцов, которые могут быть проанализированы одновременно. Он имеет преимущество, заключающееся в большом сокращении объемов реакции, требующем меньшего количества реагентов и получающемся при меньших затратах. Разработанный прибор, то есть Danaher Motion Polonator model G. 007, способен производить от 10 до 35 фунтов стерлингов за модуль за 2,5 дня работы. Прибор может соединяться с 200 из этих модулей для сбора 100 диплоидных геномов при 30-кратном покрытии за 5 дней, а оставшиеся 5 дней используются для повторения любых слабых запусков, чтобы обеспечить 98% - ное покрытие с точностью 1E-5. При значительном сокращении объема реагентов стоимость единицы объема снижается примерно в 10 раз, и компания надеется в ближайшее время достичь цели в 1000 долларов за геном.
Технология секвенирования ионных потоков
В недавнем продвижении была введена первая технология секвенирования PostLightTM (Ion Torrent) (http://www.iontorrent.com/). Эта технология создает прямую связь между химическим веществом и цифровой информацией, обеспечивая быстрое, простое и масштабируемое секвенирование. Он использует простую химию нуклеиновой кислоты Уотсона для невероятно мощной, запатентованной полупроводниковой технологии-закона Мура (Moore 1965). Принцип ионообменной полупроводниковой технологии основан на хорошо охарактеризованном биохимическом процессе, при котором нуклеотид включается в цепочку ДНК полимеразой, в результате чего в качестве побочного продукта выделяется ион водорода (рис. 2). Технологическое устройство использует массив высокоплотных микрообработанных скважин для выполнения этого биохимического процесса массово параллельным образом, причем каждая скважина содержит свой собственный шаблон ДНК. Под скважинами находится ионно-чувствительный слой, а под ним-собственный ионный датчик. Массивное параллельное секвенирование на секвенсоре Ion Personal Genome Machine (PGM ™ ) работает по принципу “базы”. Например, если к матрице ДНК добавить нуклеотид А и включить его в цепочку ДНК, то высвободится ион водорода. Заряд от этого иона изменит рН раствора и может быть обнаружен непосредственно ионным датчиком без сканирования, камер и света. Таким образом, секвенсор PGM ™ последовательно заполняет чип одним нуклеотидом за другим. Разработанная система PGM ™ позволяет выполнять широкий спектр секвенирующих приложений, таких как мультиплексирование ампликонов, транскриптом, малая РНК, а также чип-секвенирование и метилирование парных концов.
Что касается вопросов, связанных с качеством и анализом геномических данных, то значительная сумма финансирования в размере 10 миллионов долларов была предложена премией Archon Genomics X PRIZE (AGXP) для того, чтобы генерировать быстрые, точные и полные последовательности ДНК человека для глобального исследовательского сообщества (редакционная дискуссия: К медицинской последовательности генома человека. Nat Genet. 2011 Mar, 43 [3]: 173). Поскольку очень многие исследователи генома заинтересованы в этом, AGXP предлагает помощь в процессе консультаций с сообществом, чтобы помочь разработать справедливые и эффективные методы проверки данных генома конкурсантов с высокой степенью точности и полноты (Kedes et al. 2011). С момента запуска AGXP в 2006 году были достигнуты важные успехи в протоколах валидации технологий секвенирования ДНК как с точки зрения скорости, так и с точки зрения снижения затрат (Sutton et al. 2011). Однако ни одна текущая последовательность генома человека не является полностью полной, полностью точной или определенной, чтобы содержать все перестройки или информацию о фазировании хромосом (гаплотип). Сильно повторяющиеся и другие области генома остаются трудными для секвенирования, но, вероятно, будут иметь решающее значение для определения наследуемых признаков. Таким образом, идеалы X премии остаются столь же важными для будущего человеческой генетики и генетической медицины, как и прежде.
Сравнение второй и третьей платформ HT-NGS
В отличие от вторых технологий HT-NGS, которые полагаются на ПЦР для выращивания кластеров заданного шаблона ДНК, прикрепляя кластеры шаблонов ДНК к твердой поверхности, которая впоследствии визуализируется при секвенировании кластеров синтезом в поэтапном подходе, третьи технологии HT-NGS опрашивают отдельные молекулы ДНК таким образом, что не требуется никакой синхронизации (ограничение второго HT-NGS) (Whiteford et al. 2009), тем самым преодолевая проблемы, связанные с искажениями, вносимыми ПЦР-амплификацией и дефазированием. Кроме того, третьи технологии HT-NGS имеют потенциал для более полного использования высоких каталитических скоростей и высокой технологичности ДНК-полимеразы или вообще избегают какой-либо биологии или химии, чтобы радикально увеличить длину считывания (с десятков оснований до десятков тысяч оснований на считывание) и время получения результата (от дней до часов или минут). Кроме того, третьи технологии HT-NGS могут предложить следующие преимущества по сравнению со вторыми технологиями HT-NGS: i) более высокая пропускная способность, ii) более быстрое время оборота (например, секвенирование метазоанских геномов с высоким кратным охватом в минутах), iii) более длинные длины считывания для улучшения сборки de novo и обеспечения прямого обнаружения гаплотипов и даже целых хромосом, iv) более высокая точность консенсуса для обнаружения редких вариантов, v) небольшие количества исходного материала (теоретически для секвенирования может потребоваться только одна молекула) и vi) низкая стоимость, там, где секвенирование человеческого генома с высоким кратным охватом менее чем за 1000 долларов ТЕПЕРЬ является разумной целью для сообщества.
За последние шесть лет было опубликовано множество оригинальных, а также всесторонних обзорных работ, связанных как со вторым, так и с третьим поколением платформ HT-NGS. Таким образом, сравнение вторых платформ HT-NGS (Roche/454, SOLiD и Illumina) и третьих платформ HT-NGS (Helicos и Pacific Biosciences и др.) сведено в таблицу 1, иллюстрирующую сходства и различия в этих технологиях по нескольким показателям. Например, с точки зрения технологических особенностей обе платформы работают с секвенированием путем синтеза, однако Вторая платформа HT-NGS имеет дело с промывкой и сканированием многих копий молекул ДНК по сравнению с прямым физическим осмотром молекулы ДНК и ее разрешением в реальном времени (т. е. без длительных циклов гибридизации или последовательных ферментативных стадий) на третьих платформах HT-NGS. Среди других различий-секвенирование РНК, где вторая платформа HT-NGS выполняла только секвенирование кДНК, тогда как прямое секвенирование РНК в случае третьей платформы HT-NGS. Что касается анализа данных, то обе платформы имеют сложность из-за большого объема данных. На вторых платформах HT-NGS проблемы mojor-это короткие считывания, которые могут быть усложнены в алгоритмах сборки и выравнивания генома, в то время как новые проблемы обработки сигналов все еще заметны на третьих платформах HT-NGS.
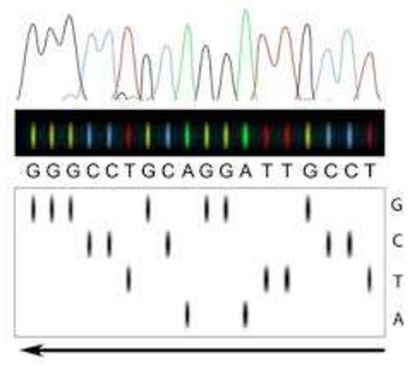
С прогрессивным появлением технологий HT-NGS затраты на секвенирование ДНК резко сократились (табл. 1). Теперь можно секвенировать сотни или даже тысячи генов для одного человека с подозрением на генетическое заболевание или сложную предрасположенность к заболеванию. Наряду с преимуществами, предлагаемыми этими технологиями, существует ряд проблем, которые необходимо решить, прежде чем широкомасштабное секвенирование станет приемлемым в практике исследований генома. Молекулярные диагносты должны будут освоиться и обрести уверенность в этих новых платформах, которые основаны на радикально отличающихся технологиях по сравнению со стандартными секвенсорами ДНК в рутинной диагностике сегодня. Начиная с 2001 года, когда появилась технология секвенирования генома человека на основе капиллярного электрофореза отдельных флуоресцентно меченых секвенирований по методу Сэнгера, появление платформ секвенирования следующего поколения резко увеличило скорость, с которой можно получить последовательность ДНК, при этом снизив затраты на несколько порядков по сравнению с их предшественниками (рис. 3). Это связано с тем, что основные механизмы генерации данных радикально изменились, производя гораздо больше последовательных считываний за один запуск прибора и при значительно меньших затратах. Полученная информация HT-NGS одновременно расширила наши знания и расширила влияние генома на биомедицинские исследования (Mardis 2011).
Эти платформы следующего поколения генерируют более короткие считывания с более низким качеством по сравнению с платформой Sanger. Сокращение длины и качества считывания потребовало разработки биоинформационных инструментов, помогающих либо сопоставлять эти более короткие считывания с эталонными последовательностями, либо собирать их de novo. Разработка этих новых методов направлена на удовлетворение спроса на информацию о последовательности в различных областях исследований, таких как изучение геномики и эволюции, криминалистика, эпидемиология и диагностика, а также прикладная терапия.
Применение и достижения технологий секвенирования в исследованиях генома человека
Веха в секвенировании генома человека была достигнута двумя группами, то есть финансируемой государством группой генома человека (HGP) и Группами Celera. Обе группы использовали разные стратегии. Группа HGP подготовила рабочий проект генома человека с помощью стратегии, основанной на карте, в то время как Celera-для секвенирования генома человека с помощью подхода whole-genome shotgun (WGS) (рис. 4). Наличие последовательного материала, полученного с помощью различных подходов, значительно облегчило способность всего научного сообщества интерпретировать полученные данные. Стратегия HGP первоначально была создана в рамках финансируемых государством усилий и основывалась на локализации бактериальных искусственных хромосом (BACs), содержащих большие фрагменты человеческой ДНК, в рамках ориентированной на ориентир физической карты. В идеале секвенирование должно было бы выполняться на основе клона за клоном, с клонами, выбранными из минимального пути укладки BAC. Ключом к стратегии HGP был последующий этап "картирования", на котором BAC были расположены на хромосомах генома путем поиска отличительных маркерных последовательностей, называемых сайтами с меткой последовательности (STSs), местоположение которых уже было точно определено. Таким образом, BACs обеспечивал карту всего генома с высоким разрешением (рис. 4). Рабочий проект, хотя и содержит некоторые пробелы и неясности, чрезвычайно полезен в таких усилиях, как идентификация генов, ассоциированных с болезнями. Одновременно идеализированная стратегия Celera состояла в том, чтобы избежать фазы предварительного картирования путем прямого субклонирования случайных фрагментов человеческого генома. Упорядочивание обоих концов фрагментов в библиотеках разного размера облегчало упорядочение. Экономя время и усилия на начальном этапе, подход Celera сделал процесс сборки гораздо более зависимым от алгоритмов и компьютерного времени. В своих попытках достичь своих целей идеализированные стратегии эволюционировали в гибриды, в которых HGP произвольно выбирал больше клонов, а Celera использовала карты BAC и последовательности, генерируемые HGP.
С момента внедрения платформы HT-NGS в 2005 году производство большого количества недорогих считываний сделало платформы NGS полезными для многих применений в исследованиях геномов человека, в частности для секвенирования генома de novo, ресеквенирования всего генома или более целенаправленного секвенирования, каталогизации транскриптомов клеток тканей и организмов (RNA-seq), обнаружения геномных вариаций и мутаций, общегеномного профилирования эпигенетических меток и структуры хроматина с использованием methyl-seq, DNase–seq и ChIP-seq (иммунопреципитация хроматина в сочетании с к ДНК– микрочипу) и персональной геномике.
De Novo, повторное секвенирование и целевое секвенирование
В целом, платформы HT-NGS сделали de novo сборку большинства организмов, включая человека, длительным и дорогостоящим процессом. У людей такая попытка уже началась с публикации нескольких полных геномов, например: с использованием технологии Roche 454 для 7,5-кратного охвата генома человека (Wheeler et al. 2008), последовательностей генома человека китайца (Wang et al. 2008), африканца (Pushkarev et al. 2009) и двух корейцев (Ahn et al. 2009; Kim et al. 2009), все они были сделаны с помощью анализатора генома Illumina и секвенированы вокруг 20-кратного гаплоидного покрытия генома, за исключением генома африканского самца, который также был повторно секвенирован в системе ABI SOLiD (McKernan et al. 2009). Совсем недавно геном Джеймса Лупски был секвенирован до 30-кратного базового покрытия с использованием твердой системы ABI (Lupski et al., 2010). Повторное секвенирование генома человека не ограничивалось платформами второго поколения. Например, геном Стивена Квейка был секвенирован до 90% покрытия генома на платформе одномолекулярного секвенирования Helicos (Пушкарев и др., 2009). Подход к генотипированию всего генома на HT-NGS эффективно обеспечивает неограниченное мультиплексирование и неограниченный выбор однонуклеотидного полиморфизма (SNP), например типирование генотипов HLA у человека (Lind et al. 2010) и геномное генотипирование плода с использованием неинвазивных HT-NGS крови матери (Burgess 2011).
Секвенирование РНК
HT-NGS также находит применение в изучении малых РНК. Например, комплексное исследование миРНК при остром миелоидном лейкозе, проведенное HT-NGS, выявило дифференциально экспрессируемые сайты связывания миРНК при остром миелоидном лейкозе (Ramsingh et al., 2010). В недавних исследованиях было введено несколько эффективных процедур для выполнения РНК-Seq с использованием платформы секвенирования Illumina (Buermans et al. 2010; Nagalakshmi et al. 2010), включая технические вопросы (Marguerat and Bähler 2010), Построение сложной базы данных репертуара миРНК (Lee et al. 2010), подготовка библиотек малых РНК и анализ полученных данных последовательностей для измерения обилия микроРНК (Morin et al. 2010), а также аннотирование и обнаружение малых РНК по транскриптомным данным (Yang et al. 2011). Было также обнаружено, что РНК seq с использованием технологий Illumina и 454 является мощным инструментом для обнаружения новых слияний генов в линиях и тканях раковых клеток (Maher et al. 2009). Понимание транскриптома необходимо для интерпретации функциональных элементов генома и выявления молекулярных составляющих клеток и тканей, а также для понимания развития и болезни. Специфическими целями транскриптомики являются: (1) каталогизация всех транскриптов в контексте типов клеток вида, включая мРНК, некодирующие РНК и малые РНК; (2) определение транскрипционной структуры генов с точки зрения их начальных сайтов, 5’-и 3’ - концов, паттернов сплайсинга и других посттранскрипционных модификаций; (3) количественная оценка уровней экспрессии каждого транскрипта в процессе развития или при различных физиологических и патологических состояниях. С появлением более быстрых и дешевых платформ HT-NGS большее количество транскриптомных анализов выполняется с использованием недавно разработанного подхода глубокого секвенирования (Wang et al. 2009). Короткие считывания, производимые технологиями HT-NGS, особенно Illumina и SOLiD, возможно, подходят для профилирования экспрессии генов. РНК-Seq был использован для точного мониторинга экспрессии генов конкретных генов для определения дифференциального сплайсинга, аллель-специфической экспрессии транскриптов и многих связанных с биологией вопросов, используемых в экспериментах с РНК-Seq (Costa et al. 2010b).
Эпигенетика
Технологии HT-NGS потенциально способны существенно ускорить эпигеномные исследования (изучение наследуемой регуляции генов, включающей не саму последовательность ДНК, а ее модификации и структуры более высокого порядка), включая посттрансляционные модификации гистонов, взаимодействие между факторами транскрипции и их прямыми мишенями, позиционирование нуклеосом в масштабе всего генома и характеристику паттернов метилирования ДНК (Bormann et al. 2010; Fouse et al. 2010; Bhaijee et al. 2011). Модификация гистонов и метилирование ДНК являются двумя важными эпигенетическими механизмами, регулирующими транскрипционный статус генов. Используя технологию ChIP-Seq (chromatin immunoprecipitation and direct sequencing), посттрансляционные модификации гистонов и расположение транскрипционных факторов могут быть изучены на уровне всего генома (Neff and Armstrong 2009), тогда как протоколы иммунопреципитации метилированной ДНК (MeDIP) и бисульфита могут быть использованы для изучения метилирования самой ДНК (Popp et al. 2010). Например, используя ChIP-seq на платформе HT-NGS, сайты связывания фактора транскрипции (TF) и связанного с ростом человека связывающего белка (GABP альфа) были непосредственно секвенированы вместо того, чтобы быть гибридизованными на чиповом массиве и распутывать широкие и сложные генные пути, регулируемые геном PPARG (Costa et al. 2010a), и предсказали открытие мотива de novo (Jiao et al. 2010). Этот чип-Seq на платформе HT-NGS теперь позволяет исследователям улучшать как количество, так и качество получаемых данных. Среди других распространенных высокопроизводительных подходов взаимодействие белок-ДНК изучалось с помощью комбинации иммунопреципитации хроматина с микрочипом ДНК (ЧИП-чип). Напротив, технология ChIP-seq наследует от платформ HT-NGs два преимущества: во-первых, она не ограничена содержанием микрочипов и, во-вторых, не зависит от эффективности гибридизации зондов. Подход ChIP-seq недавно был использован для идентификации сайтов связывания двух транскрипционных факторов, STAT1 и NRSF, в клетках человека (Robertson et al. 2007; Euskirchen et al. 2007). Оба исследования сравнивали свои результаты с результатами, полученными с помощью ChIP-chip, демонстрируя, что ChIP-seq имеет лучшее разрешение и требует меньшего количества копий.
Геномная вариация и обнаружение мутаций
NGS обещает облегчить общегеномные исследования структурных вариаций человеческой популяции (Xi et al. 2010; Henn et al. 2010), раскрыв все распространенные и редкие генетические вариации в человеческих популяциях (Bowne et al. 2011). Действительно, “Проект 1000 геномов” на сегодняшний день добился большого прогресса в достижении этой цели (Durbin et al., 2010). Имея полную генетическую карту всех человеческих вариаций, полученную NGS, исследователи смогут проводить более детальные эксперименты для выявления генетических вариаций, лежащих в основе реакции на лекарства. Патформы HT-NGS также нашли применение в высокопроизводительном обнаружении мутаций и скрининге носителей с использованием метода, называемого функциональной геномной дактилоскопией (FGF). Метод подразумевает селективное обогащение функциональных геномных областей (экзом, промотор или энхансеры сращивания экзонов) в ответ на обнаружение причинных мутаций для болезни и лекарственного ответа (Senapathy et al., 2010). Обогащение мишеней на основе микрочипов также позволило провести параллельный, крупномасштабный анализ полных геномных областей для нескольких генов патологического пути и для нескольких образцов одновременно, что обеспечило эффективный инструмент для комплексного диагностического скрининга мутаций (Amstutz et al. 2011). Скрининг носителей с помощью HT-NGS также возможен для общей популяции с тяжелыми рецессивными детскими расстройствами (Bell et al. 2011) и в обнаружении мутаций, связанных с аутосомно-рецессивной атаксией мозжечка, путем объединения анализа связей на основе SNP-массива и целенаправленного повторения соответствующих последовательностей в интервале связей (Vermeer et al. 2010).
Заключительное резюме и будущие перспективы
В быстрорастущих технологиях HT-NGS главная задача состоит в том, чтобы справиться с анализом обширной производственной базы данных секвенирования с помощью передовых инструментов биоинформатики. Поскольку 2011 год был отмечен как 10-я годовщина первого секвенирования генома человека, nucleic acid research (NAR) недавно опубликовала свой 18-й ежегодный выпуск базы данных (Том 39, дополнение 1 января 2011 года: http://nar.oxfordjournals.org/content/39/suppl_1) посвятить 10 лет достижениям в области секвенирования генома и его будущим задачам. Опубликованные выпуски состояли из 96 новых онлайновых баз данных, охватывающих различные данные молекулярной биологии, и 83 информационных ресурсов, которые ранее были опубликованы в NAR или других журналах, в общей сложности коллекция баз данных теперь включает 1330 источников данных.
Наличие сверхглубокого секвенирования геномной ДНК в ближайшем будущем преобразит медицинскую (в анализе причин заболеваний, разработке новых лекарств и диагностике) и ветеринарную (генетическое улучшение здоровья и продуктивности животных) сферы. Кроме того, он может стать перспективным инструментом в анализе иммунопреципитации хроматина в сочетании с микрочипом ДНК (ChIP-chip) или секвенированием (ChIP-seq), секвенированием РНК (RNA-seq), генотипированием всего генома, сборкой de novo и повторной сборкой генома, широкой структурной вариацией генома, обнаружением мутаций и скринингом носителей, обнаружением наследственных нарушений и сложных заболеваний человека, подготовкой библиотеки ДНК, парными концами и геномными захватами, секвенированием митохондриального генома и персональной геномикой и т. Д. Ожидается, что технология HT-NGS для клинических целей в медицине человека и внедрения геномной селекции в программы разведения сельскохозяйственных животных, вероятно, будет полностью принята в ближайшие пару десятилетий. Последние технологические достижения в области анализа HT-NGS не только ставят точку отсчета в продвижении исследований в области геномики, но и в области протеомики, других омик и исследований рака беспрецедентными темпами (например, ДНК-и белковые микрочипы, количественная ПЦР, масс-спектрометрия и другие). Хотя HT-NGS с короткой последовательностью считывания ДНК (25-50 оснований) и умеренной последовательностью считывания (500 оснований) уже нашли много потенциальных применений, но для геномного секвенирования и анализа все более важных структурных генетических вариаций в геномах, таких как вариации числа копий, хромосомные транслокации, инверсии, большие делеции, вставки и дупликации, было бы большим преимуществом, если бы длина считывания последовательности на исходной единственной молекуле ДНК могла быть увеличена до нескольких 1000 оснований и более в секунду. С развитием третьей платформы NGS огромными темпами можно надеяться, что цель определения целой хромосомной последовательности из одной исходной молекулы ДНК или последовательности генома за 1000 долларов может быть осуществлена в ближайшем будущем.
Sequencing technologies and genome sequencing | SpringerLink
Pareek, C.S., Smoczynski, R. & Tretyn, A. Sequencing technologies and genome sequencing. J Appl Genetics 52, 413–435 (2011). https://doi.org/10.1007/s13353-011-0057-x

Комментарии (0)
Зарегистрируйтесь, чтобы добавить комментарий